Published on
Urgent message: This is the urgent message about a truly urgent case presentation concerning a pediatric patient in an urgent care center.
Diana Sofia Villacis Nunez, MD; Amit Thakral, MD, MBA; and Pareen Shah, MD
CASE PRESENTATION
A 12-year-old previously healthy female presents with a 5-day history of lower extremity rash and low-grade fever (100.6°F). A month earlier, she had a self-resolving viral upper respiratory infection. The rash is described as mildly pruritic, dark red spots which started on her feet and spread upwards to her thighs. Associated symptoms include decreased appetite, bilateral ankle pain and swelling, bilateral feet swelling, and refusal to walk. Patient did not complain of abdominal pain, nausea, vomiting, headaches, dizziness, dysuria, hematuria, hematochezia, or melena.
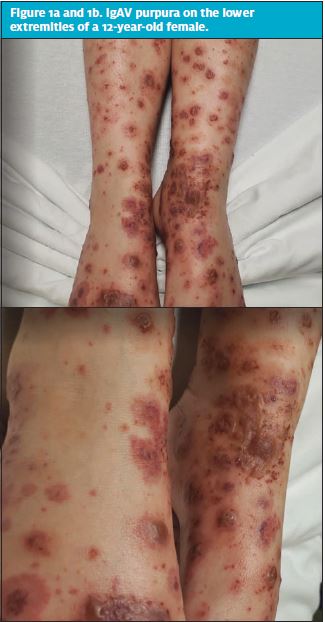
Therapy at home included ibuprofen as needed for pain and diphenhydramine for pruritus. At initial presentation in the urgent care center, she was afebrile (98.6°F) with a heart rate of 101 beats/minute, respiratory rate of 20 breaths/minute, blood pressure of 123/79 mmHg, and an oxygen saturation of 99% on room air. Physical examination showed a palpable, purpuric, non-blanching rash, most prominent on the lower legs and ankles (Figures 1a and 1b). Her ankles were notable for joint effusions bilaterally without erythema. She had full range of motion of all joints, but experienced pain with passive and active movement. Her abdomen was soft and nontender without palpable organomegaly. The rest of the physical exam was unremarkable.
OVERVIEW
IgA vasculitis (IgAV), formerly known as Henoch-Schӧnlein purpura (HSP), is the most common systemic vasculitis in childhood, affecting eight to 20 per 100,000 children each year accounting for roughly 50% of pediatric vasculitis cases in the United States.1,2 It involves small vessels with predilection for the skin, gastrointestinal tract, and kidney vasculature.3, Most cases present at between 2 and 10 years of age, with a peak incidence at 4 years of age; it is less common in teenage years and adulthood.4,5 There is seasonal variation with most cases occurring during the fall and winter seasons2 and there is a strong association with preceding or intercurrent viral disease or Group A streptococcal infection.2 IgAV is typically benign, but there are several complications that require hospitalization and/or aggressive treatment.
HISTORY
Children with IgAV classically present with a purpuric rash involving the lower extremities which may be accompanied by arthralgias, abdominal pain, and/or renal involvement. Abdominal pain and joint pain can precede the rash by up to 1 week2; this can make diagnosis difficult since the rash is necessary for the diagnosis. Musculoskeletal symptoms, however, can be delayed for up to a month after the development of rash, and nephritis can manifest several months later.6
Skin
Skin rash is visible in 95%–100% of cases at diagnosis6 and presents over the course of a few days. It usually appears in crops and may start as erythematous macules that progress into the classic purpuric rash. It can be painful and pruritic. Individual lesions tend to last 3–10 days.2 Absence of the typical rash especially, during early stages of the disease, does not completely exclude the diagnosis of IgAV.
Gastrointestinal
Gastrointestinal symptoms are present in approximately 60% of patients with IgAV.5,7 Acute, diffuse, colicky abdominal pain is the most common gastrointestinal manifestation. The intensity can range from mild to severe, prompting a surgical evaluation. Other reported symptoms can include hematemesis, melena, and hematochezia, likely indicating gastrointestinal bleeding from mucosal edema.
Musculoskeletal
An estimated 70%–90% of patients with IgAV develop arthralgias or arthritis at some point during their illness.4,7 Articular involvement has a predilection for the lower extremities, especially the knees, and ankle joints, subsequently interfering with ambulation.2 Young children generally seek medical attention due to joint swelling and refusal to bear weight.
Renal
Renal involvement can occur in 20%–80% of patients with IgAV.4,8 Most patients with IgAV nephritis are asymptomatic. However, the presence of gross hematuria, decreased urinary output, and lower-extremity edema should alert the clinician for possible renal involvement. Renal manifestations can include minor urinary abnormalities (hematuria, non-nephrotic range proteinuria), nephritic syndrome, and nephrotic syndrome, and can eventually lead to end-stage renal disease in a minority of patients.3,8
PHYSICAL EXAMINATION
Vital Signs
Low-grade fever can be expected, especially in the setting of other intercurrent illness, but high fever should prompt investigation for bacterial infection, sepsis, and disseminated intravascular coagulation. Tachycardia or hypotension may be observed with dehydration or with acute anemia due to significant blood loss from the gastrointestinal tract, although significant anemia is rare. The presence of hypertension may be a sign of renal involvement.
Skin
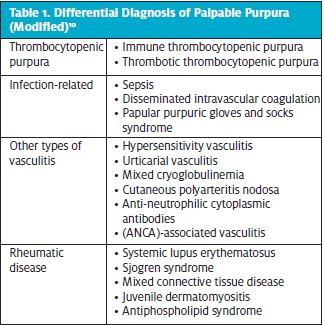
The characteristic IgAV rash is the presenting symptom for the majority of patients. It is important to expose all areas of skin to evaluate the features of the rash and the extent of involvement. The skin lesions are purpuric, non-blanchable and symmetrically distributed, and can be surrounded by skin edema and erythema. The rash is neither itchy nor painful and typically involves the buttocks and lower extremities, but it can also affect other areas of the body.2,4 Urticarial lesions can be present in some patients.9 Petechiae can also be observed. The lesions can be complicated by necrosis and bullae formation. More commonly in younger patients, non-pitting edema that involves the scalp, face, trunk, and extremities can be present.2 Scrotal swelling can also be observed, even in the absence of orchitis. The rash should be differentiated from other causes of petechiae or palpable purpura (Table 1).
Gastrointestinal
The abdominal exam of a child with IgAV is typically benign. A tender or distended abdomen warrants further investigation for GI complications, such as intussusception.3
Musculoskeletal
Patients with IgAV joint involvement typically have effusions, pain, and/or limited range of motion. These joint symptoms typically involve the lower extremities and rarely affect more than five joints.2,4
Other
Other, less common manifestations include orchitis, pulmonary hemorrhage, and central nervous system involvement manifesting as altered mental status and seizures.4
DIAGNOSIS
IgA vasculitis is a clinical diagnosis. Laboratory and other diagnostics are employed to assess for complications or other conditions that mimic IgAV; this necessity is dependent on the severity of disease.
There is no single serologic study that can confirm the diagnosis of IgAV. When the diagnosis is not obvious from history and physical examination alone, ancillary studies may be used to exclude other conditions that can mimic IgAV.
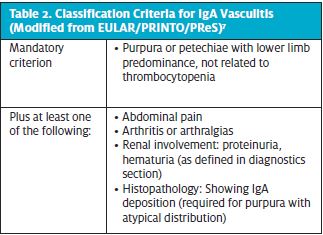
Classification criteria by the European League Against Rheumatism (EULAR), Pediatric Rheumatology European Society (PReS), and Pediatric Rheumatology International Trials Organization (PRINTO) (Table 1) provides 100% sensitivity and 87% specificity for classification of IgAV, and can serve as a guide for physicians evaluating these patients.7
EVALUATION
All patients with IgAV should be screened for associated kidney disease which may be relatively asymptomatic. This evaluation should include a urinalysis and a blood pressure measurement with an appropriate pediatric-sized cuff. Patients with significant hypertension, edema, proteinuria, or hematuria should have further assessment including urine protein, urine creatinine, and a basic metabolic panel including electrolytes and BUN/Cr. If the patient has normal urine output and no generalizable edema, this added evaluation is nonurgent and can be performed on-site or sent to a reference laboratory.
If the rash has extensive petechiae or if the dermatologic features are not diagnostic, patients should have a complete blood count with differential and a prothrombin time. These tests primarily evaluate for the absence of thrombocytopenia and coagulopathy, which are diagnostic criteria. These tests can be done non-emergently unless the patient has extensive petechiae, concern for GI blood loss, or other symptoms to suggest a bleeding disorder. If thrombocytopenia is present, causes of thrombocytopenic purpura or acute infection should be explored.10 Mild leukocytosis and thrombocytosis can be evidenced in some patients.5
Patients with IgAV arthritis do not typically require further diagnostic evaluation for the joint symptoms. If there is significant pain, high-fever, or other signs of an infectious/septic joint, further evaluation can be pursued as needed. This evaluation could include inflammatory markers (eg, CRP, ESR) or dedicated infectious testing (eg, Lyme titers).
Patients with IgAV abdominal pain may require further evaluation depending on the severity of the pain. It is helpful to test for fecal occult blood even in the absence of reported GI bleeding as it can be positive in patients without reports of bloody or melanic stools.6 Patients with significant abdominal pain should be referred to a pediatric ED for an abdominal ultrasound to evaluate for an ileo-ileal or ileo-colic intussusception.
When the diagnosis is indeterminate or if the rash distribution or characterization is atypical, a referral to Dermatology is warranted to consider skin biopsy.7 Additionally, serum IgA level elevation is seen in more than 50% of patients, which may be a helpful adjunct when there is diagnostic uncertainty.9,11
MANAGEMENT
Most cases can be safely managed in the urgent care setting. Patient transfer to a hospital should be considered depending on the severity of symptoms after consultation with a pediatric specialist. The most common reasons for transfer include severe abdominal pain, inability to ambulate, gastrointestinal hemorrhage, renal insufficiency, significant nephritis, unclear diagnosis, or (rarely) for central nervous system or pulmonary complications.
The cornerstone of management is supportive care by providing adequate hydration, analgesia, and observation. Intravenous rehydration is rarely necessary unless there is either significant abdominal pain or signs of dehydration. Skin lesions tend to have spontaneous resolution and do not require specific therapy. Pruritus, while uncommon, can be managed with oral antihistamines.
Nonsteroidal antiinflammatory drugs usually provide adequate relief for IgAV arthritis. NSAIDs should be avoided in patients with significant gastrointestinal bleeding or renal involvement. Microscopic hematuria alone is not a contraindication for NSAID use.3
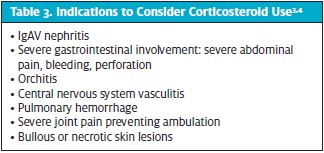
Treatment with corticosteroids should be considered in cases of severe symptoms or significant end-organ involvement in consultation with a specialist.3 Corticosteroids have not shown to alter the natural course of the disease and their use as prophylaxis to prevent development of nephritis has not been proven useful.3,4,6 The most common indications for corticosteroid use include severe abdominal pain and/or worsening arthralgias after excluding other causes. A complete list of possible indications for steroid use is offered in Table 3. The preferred corticosteroid regimen is prednisone or prednisolone 1-2 mg/kg/day for 1-2 weeks followed by a 2-4 week taper.3 Intravenous methylprednisolone at higher doses or other immunosuppressants may be used for severe, life-threatening organ involvement (gastrointestinal, lung, and CNS).3
Prognosis is overall favorable for patients with IgAV. Upon discharge, it is important to provide reassurance and education about potential complications, as well as to ensure appropriate follow-up. Patients should be evaluated by their primary care provider within 1 week. IgAV is generally a self-limited condition and resolution of symptoms typically occurs within 4–6 weeks.2
Rarely, patients can have recurrence of IgAV episodes. In these situations, it’s best to consult with a pediatric rheumatologist to consider steroid therapy for flare control.
CASE RESOLUTION
The patient’s pain and pruritus improved after one dose of ibuprofen and diphenhydramine, and she regained ambulation. She was then discharged home with ibuprofen for pain control and hydroxyzine for pruritus. Five days later, her condition worsened and she presented to the ED with leg pain and ankle swelling interfering with ambulation, intermittent mild-to-moderate abdominal pain, and a worsening rash extending up her buttocks with development of blisters. The patient was admitted to the hospital for intravenous analgesia. Oral prednisone 1 mg/kg/day was started. She did not require abdominal imaging and she did not develop nephritis. Twenty-four hours later, she was discharged home on a 6-week prednisone taper along with naproxen 250 mg twice a day for pain control with symptomatic improvement.

References
- Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schӧnlein purpura. Pediatrics. 2010;126(4):674-681.
- Reid-Adam J. Henoch-Schӧnlein purpura. Pediatr Rev 2014;35(10):447-449.
- Ozen S, Marks SD, Brogan P, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis—the SHARE initiative. Rheumatology. 2019;58(9):1607-1616.
- Oni L, Sampath S. Childhood IgA vasculitis (Henoch Schӧnlein Purpura)—advances and knowledge Gaps. Front Pediatr. 2019;7:257.
- González-Gay MA, López-Mejías R, Pina T, et al. IgA vasculitis: genetics and clinical and therapeutic management. Curr Rheumatol Rep. 2018;20(5):24.
- Jauhola O, Ronkainen J, Koskimies O, et al. Clinical course of extrarenal symptoms in Henoch-Schӧnlein purpura: a 6-month prospective study. Arch Dis Child. 2010;95(11):871-876.
- EULAR: Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schӧnlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. 2010;69(5):798-806.
- . Henoch–Schönlein purpura nephritis in children. Nat Rev Nephrol. 2014;10(10):563-573.
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain. Medicine. 2001;80(5):279-290.
- Yang Y-H, Yu H-H, Chiang B-L. The diagnosis and classification of Henoch–Schönlein purpura: an updated review. Autoimmun Rev. 2014;13(4-5):355-358.
- Trygstad CW, Stiehm ER. Elevated serum IgA globulin in anaphylactoid purpura. Pediatrics. 1971; 47(6):1023-1028.
Author affiliations: Diana Sofia Villacis Nunez, MD, Amit Thakral, MD, MBA, and Pareen Shah, MD are all affiliated with the Department of Pediatrics, Emory School of Medicine. The authors have no relevant financial relationships with any commercial interests.